When responding to a pandemic caused by a novel pathogen, such as the SARS-CoV-2 virus (COVID-19), the federal government’s basic strategy is to deploy the nation’s public health infrastructure as the first line of defense.REF The U.S. Centers for Disease Control and Prevention (CDC),REF along with state and local public health laboratories,REF are expected to collect, synthesize, and disseminate data and information related to the pathogen.REF Once the pathogen is better understood, commercial test developers and clinical laboratories can provide the capacity needed for conducting widespread testing. Non-clinical laboratories, such as those in academia, play a supporting role by conducting research to better understand the pathogen.
How COVID-19 “Stress Tested” America’s Testing System
The onset of COVID-19 proved to be a real-time stress test of this strategy. It revealed three sets of issues that inhibited a rapid response and which policymakers need to address: (1) statutory ambiguities regarding laboratory-developed tests that created uncertainty and confusion; (2) regulatory rigidities within the Clinical Laboratory Improvement Amendments (CLIA) that handicapped clinical laboratories’ response capabilities; and (3) the absence of mechanisms for leveraging the resources and capabilities of non-clinical laboratories. Those problems were further compounded by the government’s over-centralized approach to initial test development, production, and distribution.
Learning from experience with COVID-19, policymakers should reform the laws and regulations governing tests and testing to address these problems. Specifically, Congress and the U.S. Food and Drug Administration (FDA) should clarify statutory and regulatory frameworks regarding laboratory-developed tests, and the Centers for Medicare and Medicaid Services (CMS) should revise regulations both to provide clinical laboratories with greater flexibility to respond to emerging threats and situations and to provide qualified non-clinical laboratories and personnel the opportunity to utilize their testing expertise for clinical purposes. Federal policymakers should also make permanent a number of changes made to regulatory policies during COVID-19 that provided additional flexibility.
The government’s oversight of clinical testing needs to be reformed to accommodate more decentralized, streamlined, and innovative approaches—and to remove inadvertent regulatory impediments.
Overview of Testing
Broadly speaking, researchers and clinicians perform tests on biological samples (i.e., tissue, blood, saliva, hair, and urine, etc.)REF to collect, interpret, and evaluate data related to basic research, applied research, and medical care.REF Any given test consists of both its physical components and its protocol. The components include reagents (substances involved in chemical reactions) and any associated single-use supplies or multi-use equipment. The protocol is a set of specific instructions for using the components to perform the test and interpret the test results. Testing is conducted by trained personnel in various facilities, generally called laboratories, most of which fall into one or more of four broad groups: governmental, commercial, clinical, or academic.
The same test can be used for different purposes. For instance, a test that measures cholesterol levels could be used in the context of basic research to determine the effects of cholesterol on cellular properties, in the context of applied research to determine the effectiveness of a new drug to regulate cholesterol, or in the context of medical care to determine a patient’s risk of developing heart disease.
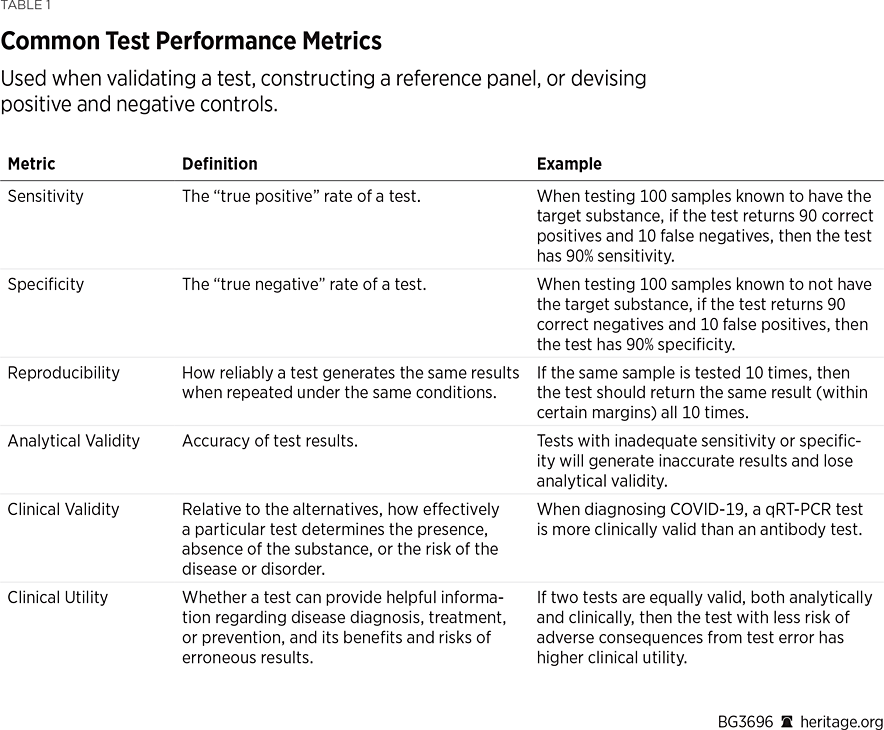
When a new test is developed, it must be validated using positive and negative controls. Positive controls are samples known to contain the target substance being tested for, while negative controls are known to not contain the substance. These controls are used to assess test performance and can also be used to assess the relative performance of two or more tests for the same substance. Table 1 summarizes key metrics used to validate a test. Once a test is appropriately validated, it can then be used with confidence that it will yield the desired information.
The Regulation of Clinical Tests and Clinical Testing
Testing can be done for either research purposes or clinical purposes (i.e., patient care), and is subject to a number of different laws and regulations. Some, such as those governing the safe use of hazardous materials, are universally applicable.REF Non-clinical testing is also subject to standards and requirements related to ethical considerations when conducting research on animal or human subjects.REF In the case of clinical testing, both the tests used and the facilities and personnel performing the tests are subject to regulation, but under two different statutes administered by two separate federal agencies. That long-standing division of authority and responsibility, while logical in some respects, has inadvertently created points of ambiguity and regulatory uncertainty, which were factors that contributed to America’s delayed testing response to the COVID-19 pandemic.
Two main areas of federal law and regulation apply to testing done for clinical purposes. One consists of a set of provisions within the Food, Drug, and Cosmetic Act (FD&C) that applies to tests marketed for clinical use, and which is administered by the FDA.REF The other is the Clinical Laboratory Improvement Amendments of 1988, which applies to the personnel performing clinical tests and the facilities (laboratories) in which they work, and which is administered by the CMS.REF
While conceptually these are two separate and distinct regulatory regimes, there are instances where they overlap—resulting in statutory ambiguity and confusing regulations.
Regulation of Clinical Tests. Laboratory tests intended for use in disease diagnosis, treatment, or prevention are considered “in vitro diagnostics.” Under the federal FD&C, in vitro diagnostics are classified as medical devices subject to FDA regulation, including pre-market analytical and clinical validity standards, pre-market approval, and post-market surveillance and adverse-event reporting requirements.REF Historically, these standards have been differentially applied to commercial tests and laboratory-developed tests. Commercial tests are developed with the intention of being marketed and distributed to, and used within, multiple laboratories. These tests are classified and regulated according to their relative complexity and risk of adverse consequences from erroneous results as Class I, II, or III, and their manufacture and marketing is subject to FDA medical device approval and regulatory requirements.REF
Once the FDA has approved a commercial test, it assigns it a “low,” “moderate,” or “high” complexity rating. If a moderate complexity test is simple and unlikely to generate an incorrect result, a test developer can apply for it to be classified as low complexity. These ratings also factor into CMS regulation of clinical laboratories and testing personnel under CLIA, as they are also used as the basis for determining the kinds of tests that any given clinical laboratory is permitted to perform and for defining the required proficiencies of testing personnel.REF
In contrast, “laboratory-developed tests” are developed by a single laboratory with the intent of being used exclusively within that laboratory. There are four basic categories of laboratory-developed tests:
- Tests that are created entirely in-house with components and reagents assembled—and sometimes produced—within the laboratory. These are informally referred to as “home-brew tests.”
- Tests that a laboratory develops in accordance with protocols developed by another laboratory.
- Tests for which the laboratory uses a commercial test but substitutes one or more components or reagents with ones made in the laboratory or sourced from a different vendor.
- Tests that modify the protocol of an existing test, but do not otherwise alter or substitute any components or reagents.REF
Under current regulations, all laboratory-developed tests are classified as “high complexity” by default. However, the FDA has historically exercised enforcement discretion to exempt most laboratory-developed tests from pre-market review requirements. This occurred because laboratory-developed tests are generally not marketed or widely distributed, and originally they had limited and simple applications. In recent years, as laboratory-developed tests have become more commonplace and technically complex, the FDA has determined that they warrant greater oversight.
In 2014, the FDA published draft guidanceREF regarding regulation of laboratory-developed tests but announced in 2016 that they would be delaying its finalization. This has since left laboratory-developed tests in a problematic and murky regulatory landscape.
Regulation of Clinical Testing Facilities. Labs that use in vitro diagnostics for clinical purposes, including both commercial tests and laboratory-developed tests, are considered clinical laboratories subject to the Clinical Laboratory Improvement Amendments of 1988.REF CLIA began as the Clinical Laboratory Improvement Act of 1967,REF which primarily pertained to hospital and independent labs. When it was amended in 1988, Congress expanded its scope to include all labs performing clinical testing. CLIA provides the Secretary of Health and Human Services (HHS) broad discretion in implementing regulations regarding certification of clinical labs. HHS has assigned responsibility for different aspects of CLIA to the CMS, the CDC, and the FDA.REF
To become CLIA-certified, labs must describe the number, complexity, and types of tests being performed, meet facility and personnel qualification requirements, and make records available to HHS.REF They must also undergo inspections and perform proficiency testing measures demonstrating their test performance capabilities, as well as meet any additional state-level requirements. Labs are held to additional standards if they perform specialty or subspecialty testing (e.g., bacteriology, virology, or routine chemistry).
There are also more stringent qualification requirements for personnel who perform high-complexity tests, relative to those exclusively engaged in performing low-complexity testing. As of January 2022, there were 323,086 CLIA-regulated labs, with the most common (40 percent) being physician office laboratories. Other CLIA-regulated laboratories include government-run public health labs, commercial labs, hospital labs, point-of-care sites, and academic laboratories serving clinical roles.REF
Importantly, laboratories using in vitro diagnostics for non-clinical purposes are not subject to CLIA. This means that different labs can use the same test, performed in the exact same manner, using the same type of samples, but the application of CLIA regulations to the laboratory will depend on whether the test was done for clinical or non-clinical purposes.
One effect of that distinction has been uncertainty and confusion with respect to the regulation of laboratory-developed tests. Specifically, because CLIA regulation is linked to the purpose for which testing is performed, laboratories and laboratory associations have long argued that laboratory-developed tests should be treated as services regulated by the CMS under CLIA—as opposed to being treated as products regulated by the FDA. As noted, the FDA has not finalized specific guidance on laboratory-developed test regulation, and Congress has so far failed to clarify the ambiguities entailed in applying the two statutes to laboratory-developed tests.REF
Four Problems Exposed By COVID-19’s “Stress Test” of America’s Testing System
The COVID-19 pandemic revealed four key problems regarding the regulation of clinical laboratory tests and testing that contributed to America’s delayed testing response.
Problem One: Confusing and Vacillating Regulation of Laboratory-Developed Tests. The Secretary of HHS formally declared COVID-19 to be a public health emergency on January 31, 2020.REF On February 4, 2020, the Secretary also invoked provisions in the FD&C that authorize the FDA to grant emergency use authorizations (EUAs) to diagnostic tests prior to completing the normal review and approval processes.REF
That triggering of the FDA’s authority to issue EUAs cut two ways. While it generally helped expedite the availability of commercially developed diagnostic tests, REF it paradoxically introduced increased regulatory rigidity for laboratory-developed tests. As noted, the FDA has typically exercised enforcement discretion regarding those tests, waiving pre-market review requirements. However, during public health emergencies, the FDA has applied EUA approval standards to both commercial tests and laboratory-developed tests.REF The real-world effect of that decision was that a laboratory that had developed its own COVID-19 test was prevented from using it—even in-house—without first navigating the FDA approval processes to obtain at least an EUA for its test.
This was particularly detrimental to America’s early emergency response efforts, when public health laboratories found that they were unable to validate the CDC-developed COVID-19 tests due to contamination issues.REF Because those tests were functionally useless, nearly all U.S. COVID-19 diagnostic testing had to instead be performed at CDC labs for several weeks during February of 2020 (the initial outbreak).REF Yet public health labs that modified the flawed CDC test to address its deficiencies were not allowed to use their improved versions without first obtaining FDA approval, as their modifications would be regulated as “new” laboratory-developed tests.REF Requiring approval for use of laboratory-developed tests also prevented hospital laboratories from developing their own COVID-19 tests for in-house use—something that is common practice for other diseases.
On February 29, 2020, the FDA granted the New York Public Health Laboratory an EUA for its COVID-19 test. At the same time, the FDA implemented a notification process that permitted pre-approval use of certain validated tests by high-complexity CLIA labs.REF Those decisions resulted in a rapid increase in the number of tests performed each day.REF (See Chart 1.)
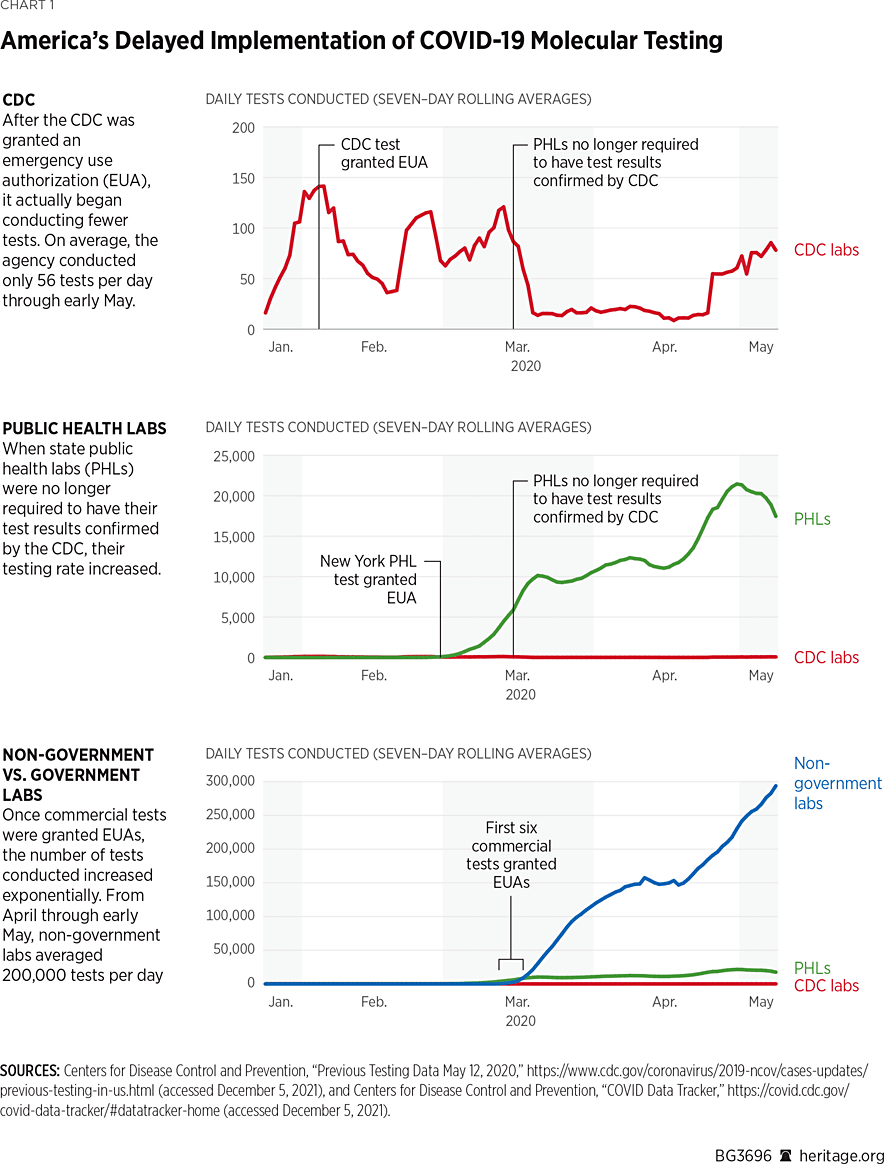
Despite this positive impact of expanding testing capacity and requests from both the Association of Public Health Laboratories and the American Association for Clinical Chemistry that the FDA return to its previous policy of exercising enforcement discretion, the agency did not rescind its new requirement for emergency use authorization of laboratory-developed tests.REF However, a legal review by the HHS Office of the General Counsel concluded that pre-market review requirements (including EUAs) for laboratory-developed tests needed to either be promulgated through notice-and-comment rulemaking by the FDA or required by an act of Congress.REF Therefore, on August 19, 2020, HHS announced that the FDA did not have the authority to require pre-market review of laboratory-developed tests absent formal rulemaking or statutory change—thus reversing the FDA’s imposition of pre-market review requirements on laboratory-developed tests.REF
Yet that HHS announcement served as only a temporary fix to decades of confusion surrounding the regulation of laboratory-developed tests. Indeed, one year later (November 15, 2021) the Biden Administration reversed that decision, holding that the FDA does have authority to require pre-market review of laboratory-developed tests absent notice-and-comment rulemaking or an act of Congress.REF This history of vacillating regulatory requirements for laboratory-developed tests signals a need for Congress to provide statutory clarity—not only for emergency situations, but for normal circumstances as well.
Problem Two: CLIA Rigidities Handicapping Response Capabilities. CLIA regulations, while put in place in good faith to protect patient health and ensure the integrity of clinical laboratory testing, created rigidities that compounded delays in testing-capacity expansion during the COVID-19 pandemic.
For instance, CLIA regulations require that each site at which clinical work is performed be certified. This means that regardless of the qualifications of laboratory personnel and the appropriateness of off-site facilities and equipment, clinical work cannot be done unless the site where it is performed is also a CLIA-certified laboratory. Thus, COVID-19-initiated lockdowns precluded testing personnel working remotely—either at home or at an ancillary facility—until the CMS began exercising enforcement discretion regarding such circumstances.REF The problems were that CLIA regulations did not anticipate the need to rapidly expand testing through temporary sites and did not account for the fact that simple tasks (such as reviewing and reporting data) can be performed safely and accurately off-site.
Another regulatory rigidity was the default application of the “high complexity” classification to all laboratory-developed tests and any other test that has not yet received FDA approval (i.e., at least an EUA). That precluded many CLIA-certified laboratories performing unapproved tests—even if the tests are actually of only “low” or “moderate” complexity.
Further compounding the situation was widespread uncertainty regarding the regulatory definition of a laboratory-developed test. Interviews conducted by The Pew Charitable Trust demonstrate that clinical laboratory personnel, including seasoned laboratory managers, were not aware that modifications to an FDA-approved test—such as deviating from the test’s protocols, substituting components or reagents in a test kit (even with ones sourced from FDA-approved vendors), or creating tests composed entirely of FDA-approved components—are all treated as the creation of a new laboratory-developed test, and thus are assigned a default “high complexity” rating under CLIA regulations.REF
As a practical matter, that meant that low- and moderate-complexity CLIA-certified labs were not permitted to use their own tests, even if their test involved only reasonable and modest modifications to an existing, FDA-approved low- or moderate-complexity test. This contributed to the issuance of over 100 cease-and-desist orders by the CMS between August 12, 2020, and October 9, 2020, to laboratories on the grounds that they were performing tests outside the scope of their certification.REF These problems were already present in CLIA’s regulatory framework but were made more visible and pressing by the need to respond quickly, and at scale, to the COVID-19 pandemic. That experience highlights another area in which Congress needs to provide additional statutory clarity.
Yet another regulatory rigidity is that CLIA regulations only permit approved tests to be used for purposes explicitly listed in their authorization notice. While that policy is intended to prevent misuse of tests and incorrect interpretation of results, it also has unintended consequences. A good example was the delayed detection of what may have been the first case of community spread of COVID-19 in the United States in February 2020.REF At that time, the CDC test was only approved for use on symptomatic individuals who had traveled to the U.S. from China (and their close contacts).REF The CMS later began exercising enforcement discretion for these situations, allowing use of approved tests for testing of asymptomatic individuals, outside the tests’ authorization, but only after cases such as this went undetected, worsening the spread of COVID-19.REF
In sum, overly restrictive CLIA regulations prevented remote performance of clinical testing work (including simple tasks such as data review), slowed establishment of temporary testing sites, precluded low- and moderate-complexity CLIA-certified labs from performing certain tests within the expertise of their staff, and prevented early detection of COVID-19 in asymptomatic individuals. Taken together, these rigidities evidence the need to reform CLIA to provide clinical laboratories with greater freedom to adapt to unanticipated situations and perform testing commensurate with their skills and capacity.
Problem Three: Preventing the Leveraging of Non-Clinical Laboratory Capacity and Resources. At a time when the U.S. urgently needed to expand testing capacity, non-clinical laboratories that sought to offer their testing expertise were sidelined by government regulators’ outdated practices and poor communication. CLIA regulations prevent any non-certified laboratory from performing diagnostic testing, even though both clinical and non-clinical laboratories often use the exact same techniques and tests. For instance, it is estimated that research facilities in America possess between 9,975 and 23,460 qRT-PCR, or digital thermocyclers—which are the machines needed to run molecular COVID-19 tests.REF Yet the CMS sent over 50 cease-and-desist orders to non-CLIA-certified labs performing unapproved testing.REF
Attaining CLIA certification during the COVID-19 pandemic, particularly to perform high-complexity testing, proved to be a significant regulatory barrier. Many labs found certification processes to be overly nuanced and confusing.REF In fact, a survey of roughly 4,000 National Institutes of Health–funded researchers conducted in early 2020 found that nearly 1,600 respondents said that they were capable of performing COVID-19 testing but were not doing so. Ninety-five percent of those respondents said that it was due in part to lack of information on protocols and regulations.REF Respondents also found the CLIA certification process antiquated and difficult to navigate.REF For instance, prior to the CMS streamlining certification processes on September 25, 2020, labs were unable to pay certification feesREF online, and once approved could not begin performing testing until they received their physical CLIA certificate in the mail.REF
Further compounding the situation was a lack of clear communication with labs and test developers by the CDC and the FDA—problems that the Government Accountability Office had previously identified in the government’s response to the bloodborne Zika virus, but which became more evident in the context of responding to a rapidly spreading airborne virus, SARS-CoV-2.REF
To unlock some of this underutilized testing capacity, the CMS began exercising enforcement discretion for what it described as “surveillance testing” by non-CLIA-certified labs.REF However, the solution devised by the CMS diverged from true surveillance testing and could more accurately be called “referential testing.” The distinctions are as follows:
- In “clinical testing,” the results are linked to the tested individual and reported to that individual or to the provider that ordered the test.
- In “surveillance testing,” the results are not reported to test subjects or health care providers. Instead, they are “de-identified” and analyzed collectively at the group or population level.REF The Seattle Flu Study was a classic example of “surveillance testing,” in which researchers collected thousands of samples to help model how seasonal influenza spreads in the population.REF Such testing is CLIA exempt.REF
- In the CMS hybrid design of “referential testing,” samples are tested for COVID-19, but are not required (as is the case with surveillance testing) to be analyzed at the group or population level. The testing facility is still, however, prohibited from reporting results to either the individuals themselves or to health care providers, and instead can only refer individuals with presumptively positive or inconclusive results for follow-up “clinical testing” at a CLIA-certified laboratory.REF
This exemption allowed non-CLIA-certified labs to perform “pooled testing” (pooling multiple individuals’ samples together and performing a single test on the pool) and to refer all individuals within a positive-testing pool for confirmatory “clinical” testing.REF The exemption, however, also allowed non-CLIA-certified labs to perform testing beyond the limits imposed on true surveillance testing, such as through “double pooled testing” and “next generation sequencing,” methods that allowed individuals who tested positive to be more easily identified. It also created institutional confusion, with some universities refusing to allow their non-CLIA-certified laboratories to perform “referential testing.”REF
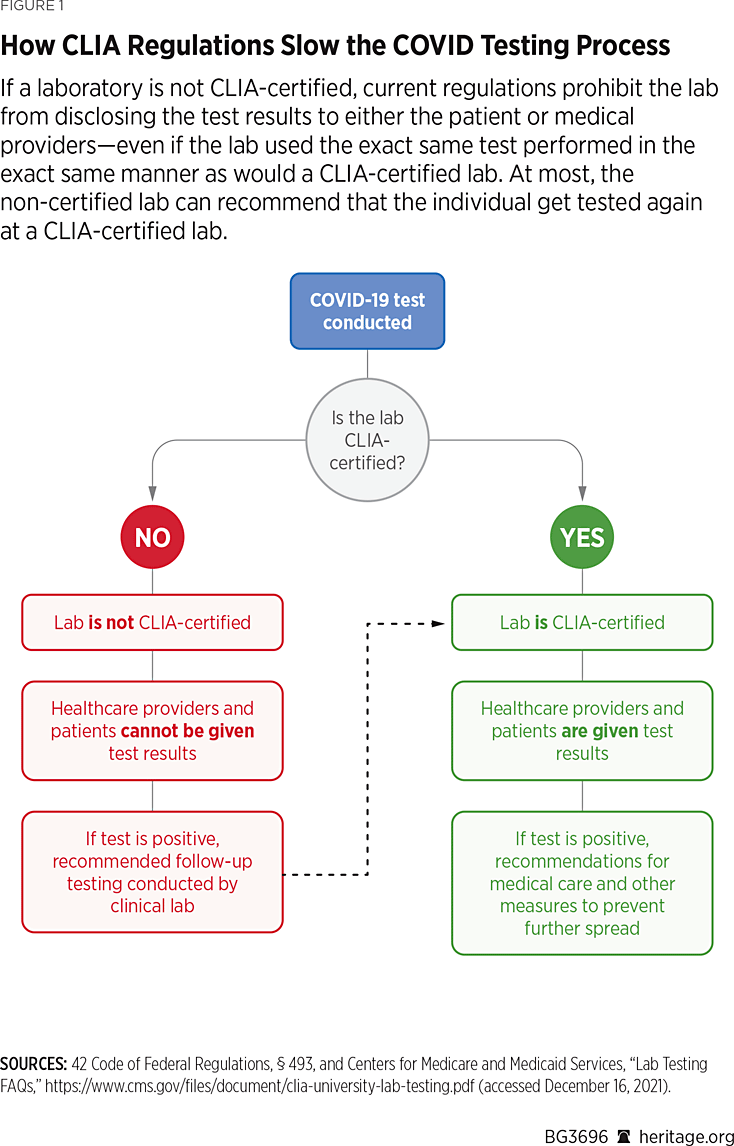
Crucially, under the CMS “referential testing” construct, non-CLIA-certified labs were still prohibited from providing diagnostic test results to patients or their health care providers. In practice, this meant that a non-CLIA-certified lab could obtain an individual’s consent and test that individual for COVID-19 (even using an FDA-approved test) but could not provide the individual with the test results. The most it could do would be to recommend that the individual get tested again at a CLIA-certified lab. As Figure 1 illustrates, these regulations resulted in unnecessary double-testing, which wasted time and resources and potentially enabled further viral spread during the interval between an individual’s “referential” test and confirmatory “clinical” test.
These problems were magnified by clinical laboratory staff shortages that have continued throughout the pandemic. As laboratory personnel have also become infected with COVID-19, clinical laboratories have been forced to limit the number of appointments, close testing sites, or otherwise reduce their testing volume.REF However, due to regulatory hurdles, non-clinical laboratories are unable to fill this need and augment clinical laboratory testing capacity.
In sum, CLIA’s regulatory distinctions between clinical and non-clinical testing effectively sidelined potential testing capacity, and CMS attempts to devise regulatory “work arounds” were largely ineffective. That experience reveals yet another area in which Congress should enact statutory reforms to enable research laboratories and their trained personnel to supplement America’s clinical testing capacity.
Problem Four: Over-Centralization Delaying Initial COVID-19 Testing. America’s testing response to COVID-19 was also delayed by an initial reliance on a single source for test development, production, and distribution—further compounded by that source being a federal government agency, the CDC.REF
In contrast, America’s remarkably rapid development and deployment of COVID-19 vaccines resulted from policymakers applying the exact opposite approach. In the case of diagnostic test development and production, the CDC relied on its internal Biotechnology Core Facility Branch Lab to manufacture test components and controls, rather than sourcing them from private companies—which the agency claimed would take longer.REF In the case of vaccine development, the primary drivers were private researchers and drug makers. Rather than supplanting their efforts, the FDA collaborated with the private sector to standardize and streamline regulatory review and approval processes, and the federal government contracted in advance to buy large quantities of vaccines if they were proven safe and effective.
Thus, the CDC’s organizational culture of top-down emergency response served to sideline innovation by state public health laboratories and the private sector—hampering the ability of other test developers and laboratories to apply their expertise to combating the pandemic.
Key Considerations and Recommendations for Policymakers
Some of the problems with testing revealed by COVID-19 resulted from previous efforts by Congress and federal regulators to ensure test quality and safety, which inadvertently created regulatory impediments to quickly responding to an emergency. In response to the pandemic, federal agencies eventually acted to amend policies and guidance to provide additional flexibility.REF However, the effectiveness of those changes was constrained by the fact that agencies were forced to make changes and exercise enforcement discretion in real time. As a result, some of their regulatory “workarounds” were suboptimal and delayed.
Learning from the experience with COVID-19, legislators and policymakers should focus on reforming the laws and regulations governing tests and testing to achieve the objectives of:
- Providing both clinical and non-clinical laboratories with increased flexibility to respond to new needs and changing circumstances;
- Streamlining and clarifying government oversight of tests and testing to make regulatory requirements and processes more transparent and rational;
- Creating mechanisms to utilize the latent ability of research laboratories to augment existing clinical testing capacity; and
- Promoting innovation, collaboration, and flexibility in developing and deploying tests for novel pathogens.
Recommendations for Establishing a Framework for Laboratory-Developed Tests
Clarify Regulatory Authority Over Laboratory-Developed Tests. In practice, current laws and regulations do not appropriately address laboratory-developed tests. The FDA has long held that it has regulatory authority over them, while others have argued that they should be considered clinical services regulated by the CMS. The FDA currently has regulatory authority over in vitro diagnostics, and under CLIA, the CMS ensures that labs meet analytical validity standards for test methods.REF Congress needs to clarify the situation to eliminate regulatory confusion.
- Congress should clarify that CMS authority under CLIA is limited to regulating the personnel and facilities performing clinical testing, while the FDA is responsible for ensuring that the tests themselves—including laboratory-developed tests—are safe and effective. It makes no sense to differently regulate in vitro diagnostic tests based on who developed the test.
- Pending such statutory clarifications, HHS should reverse its recent policy change and restore the previous Trump Administration decision to disallow the FDA from requiring pre-market review of laboratory-developed tests absent notice-and-comment rulemaking processes. The FDA should then, through notice-and-comment rulemaking, propose a better approach to regulate laboratory-developed tests, preferably along the lines of that recommended in the next section.
Devise Rules for Laboratory-Developed Tests That Avoid Hindering Innovation and Medical Care. The category “laboratory-developed tests” currently encompasses a range of possible tests, many of which are really “laboratory-modified tests,” in that they are not truly novel tests but rather modified versions of existing tests. To avoid stifling innovation and access to medical care, the applicable statutes and regulations should be revised to more appropriately accommodate relevant distinctions. In general, Congress should amend the FD&C Act to:
- Provide for minimal review and near-automatic approval if a laboratory substitutes test components or reagents sourced from another FDA-approved supplier. Such situations are analogous to a drug manufacturer sourcing bulk chemicals from a different FDA-regulated supplier.
- Implement a “file and use” approach to regulating laboratory modifications to the components or protocols of an approved test. Allowing the use of test modifications during the interim period between validation and ultimate FDA review would ensure that time-sensitive medical care and innovation are not held up by onerous, costly, and time-consuming approval processes.REF
- The FDA should build upon this statutory framework and devise a risk-based approval pathway that exempts low-risk and urgently needed test modifications from pre-market review and expedites approval of non-exempt, higher-risk modifications.REF
- For truly novel laboratory-developed tests, the FDA should determine the test’s complexity when the application is filed and provide for expedited approvals of tests that are of low or moderate complexity. Novel laboratory-developed tests address unmet medical needs, respond to rapidly evolving scientific findings, and can prevent critical delays in medical care prior to commercially marketed in vitro diagnostics becoming available.
- Congress should provide the FDA with a statutory framework for expediting reviews of or implementing “file and use” approaches for certain novel laboratory-developed tests, such as ones for low-prevalence conditions.REF
Provide Mechanisms for Non-Commercial Sharing of Laboratory-Developed Tests. Commercial tests are developed with the intention of being widely marketed, distributed, and used, while laboratory-developed tests are created with the intention of solely being used within one laboratory. As mentioned previously, if a lab develops a test in accordance with the protocols developed by another lab (i.e., non-commercial sharing), it currently constitutes a new laboratory-developed test because it will be used in a different lab than the initial developing lab. To encourage interlaboratory collaboration and discourage duplicative test creation (and associated regulatory and logistical burdens), the FDA should introduce mechanisms through which laboratory-developed tests can easily be shared with other laboratories.
- Congress should stipulate that regulatory frameworks devised by the FDA appropriately consider risk mitigations inherent in the limited distribution and application of laboratory-developed tests in comparison to commercially marketed in vitro diagnostics.REF
- The FDA should create a pathway through which laboratory-developed tests can be approved for use by additional laboratories, provided the test is not commercially marketed.
Recommendation for Reforming CLIA to Expand Scope of Practice
Expand the Scope of Practice of Low- and Moderate-Complexity Clinical Laboratories. As the COVID-19 pandemic showed, providing laboratories with greater regulatory flexibility regarding CLIA requirements increased access to testing. However, the need for regulatory flexibility is not limited to only emergency situations. Ongoing innovations in medical care will continue to drive demand for clinical testing and new tests. One way that increasing demand for other medical services has been accommodated is by revising restrictions on scope of practice to enable providers to practice at the so-called top of their license.REF The CMS should similarly revise CLIA rules regarding scope of practice for clinical laboratories and testing personnel.
Specifically:
- The CMS should amend CLIA regulations to allow low- and moderate-complexity CLIA-certified labs to demonstrate their ability to safely and effectively perform a limited number of moderate- or high-complexity tests. This would also allow laboratory directors and testing personnel at those labs to utilize specific expertise they may have in order to offer certain tests classified as higher complexity than their lab’s facility certification currently allows.REF
- The CMS should revise CLIA personnel requirements by implementing an approach under which clinical laboratory directors would be responsible for ensuring that their testing personnel are capable of performing the offered testing, regardless of complexity, based on a combination of personnel proficiency evaluations, adverse-event reporting, and inspections.REF
Recommendation for Leveraging the Resources and Capabilities of Non-Clinical Laboratories
Create CLIA-Certification-Equivalent Pathways for Non-Clinical Laboratories and Researchers. The COVID-19 pandemic revealed that the U.S. needs a mechanism through which the expertise of non-clinical laboratories and researchers can be leveraged to bolster clinical testing capacity. To accomplish this, the CMS should create pathways for granting non-clinical laboratories and their testing personnel CLIA certification equivalency. Non-clinical researchers already demonstrate their technical expertise through online training and certification programs. The CMS should build on that existing framework so that those laboratories and personnel can similarly demonstrate their clinical testing capabilities.
Specifically:
- The CMS should devise online training programs and make them available on well-established government training portalsREF encompassing a set of capability assessments and personnel qualifications relevant for clinical testing.REF Individuals who complete the requisite training could then be included in a database through which they could be recruited to address temporary clinical staff and testing-capacity shortages.REF
- The CMS should amend CLIA regulations so that research laboratories whose directors and applicable testing personnel have completed the requisite training are considered CLIA-equivalent.REF Since such labs would likely perform diagnostic testing only infrequently and to a limited extent, they should only be required to perform proficiency testing challenges for the specific clinical tests they offer and should not be subject to all of the inspections, fees, and other requirements imposed on fully CLIA-certified clinical laboratories.
Recommendation for Retaining and Building Upon Regulatory Flexibilities
Making Permanent Changes That Increased Regulatory Flexibility. The lack of flexibility in the pre-pandemic regulatory framework served to delay timely adaptations needed to effectively respond to the challenges posed by COVID-19. While regulatory changes made in response to the COVID-19 pandemic helped alleviate problems, they generally provided delayed, limited, or retroactive flexibility. Even so, some of those regulatory changes should be retained to provide laboratories with more flexibility not only to respond to emerging threats, but also to function more effectively under non-emergency circumstances.
Specifically:
- The CMS should continue to allow online payment of certification fees and submission of applications, permit labs to begin testing upon CLIA identification number assignment, allow laboratories to utilize temporary testing sites and extend existing CLIA certifications to those sites, allow individualized quality control plans, allow accreditation organizations to conduct remote surveys, revise overly rigid testing criteria, and allow the use of expired test supplies in instances of shortages when quality control assurances are in place.
- The FDA should maintain the mechanisms that it put in place to better communicate with test developers and manufacturers, including mailboxes for online EUA application submissions and medical device shortages, the updating of various EUA application templates, and its creation of mechanisms to address questions and provide feedback such as a 24/7 hotline, Frequently Asked Questions, and hosted webinars and town halls.REF
- The FDA should build on those mechanisms by publishing additional templates for all test formats and specific device types and by constructing a fully online test submission portal populated with templates, applicable agency guidance, and a database containing information on all approved laboratory tests, including detailed protocols provided by the developers, any approved modifications to the tests, relevant recalls, and other pertinent information.
- The FDA should reverse its policy put in place on November 15, 2021, which re-introduced regulatory rigidities, by no longer allowing use of tests between validation and approval.REF
- As previously noted, HHS should reverse the recent rescission of the Trump Administration’s policy decision disallowing the FDA from requiring pre-market review of laboratory-developed tests absent notice-and-comment rulemaking processes.
Additional Recommendation: Shifting CDC Culture and Priorities Toward Innovation-Focused Emergency Responses
The CDC’s initial COVID-19 testing failures were largely the result of that agency prioritizing its own development and production of tests using its internal staff and facilities. The private sector is much better positioned to tackle the challenges inherent in developing and manufacturing novel products, as illustrated by the success of the FDA’s alternative approach to facilitating the development of COVID-19 vaccines and therapeutics by private companies.
When it comes to testing, the role of the CDC should similarly be to facilitate—rather than supplant—the efforts of private test developers, academic laboratories, state public health laboratories, and clinical testing providers. When responding to a novel pathogen, the CDC should focus on gathering and disseminating information, including specimens needed for development of positive controls and reference panels, and ensuring that test developers can effectively develop and validate diagnostic tests.
These changes will require a shift in priorities and culture at the CDC—and throughout HHS more broadly. HHS should revert to the more pro-innovation culture and direction of the Trump Administration that allowed atypical but successful endeavors such as Operation Warp Speed to thrive. These attitudes will be necessary to implement similar approaches to diagnostic testing at the CDC.
Conclusion
The COVID-19 pandemic proved to be a real time stress test of America’s clinical testing system, and it revealed a number of flaws and inadequacies that need to be addressed. The most notable ones are the confusing and vacillating regulation of laboratory-developed tests, rigidities in CLIA regulations that handicap clinical laboratories’ response capabilities, and the lack of mechanisms for leveraging the capabilities and personnel of non-clinical laboratories. Learning from the COVID-19 experience, policymakers should focus on reforming the laws and regulations governing tests and testing to provide greater flexibility, closer collaboration, improved communication, and better leveraging of available resources.
The good news is that the COVID-19 pandemic also demonstrated that America’s private biomedical sector is capable of rapid response and innovation in all phases of the process—from research to development to mass production. This is true not only for vaccines and therapeutics, but also for clinical tests. The job for policymakers is to ensure that laws or regulations intended to ensure the safety and efficacy of biomedical products—including clinical tests—do not needlessly or inadvertently block or delay such beneficial innovation.
Zachary B. Sluzala is a Heritage Foundation Graduate Fellow in Health Policy in Domestic Policy Studies, of the Institute for Family, Community, and Opportunity, at The Heritage Foundation. Edmund F. Haislmaier is the Preston A. Wells, Jr. Senior Research Fellow in Domestic Policy Studies.



